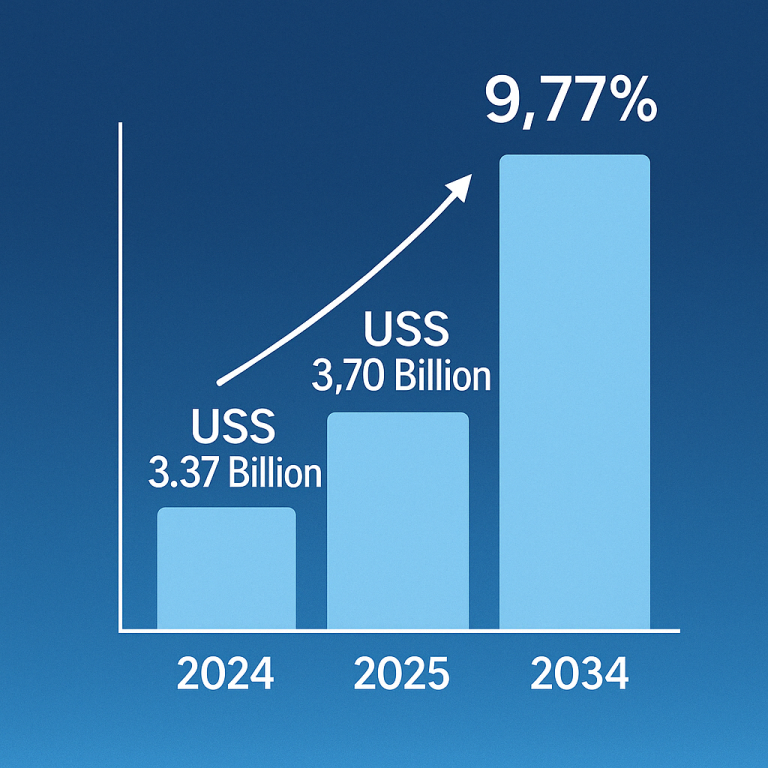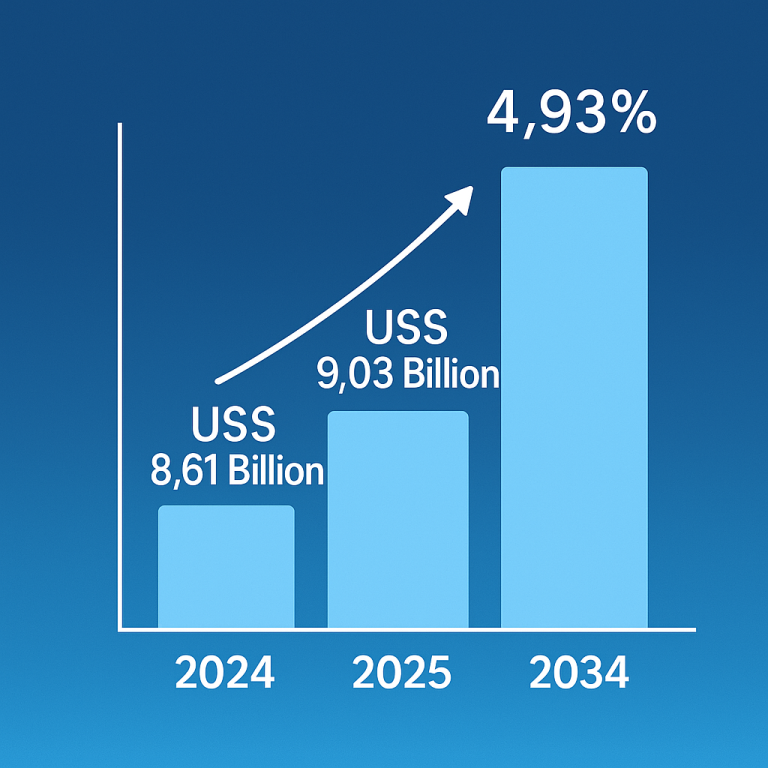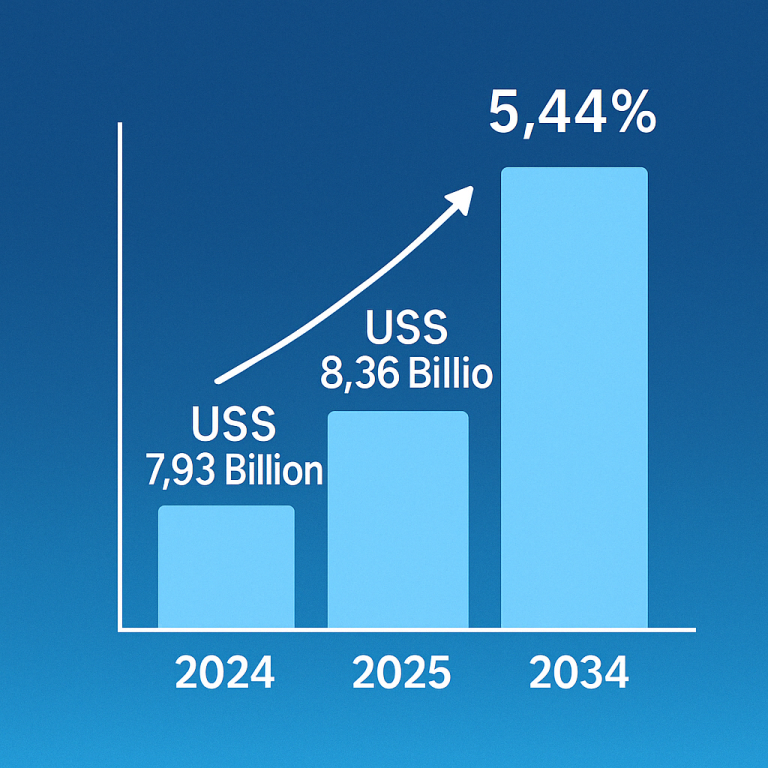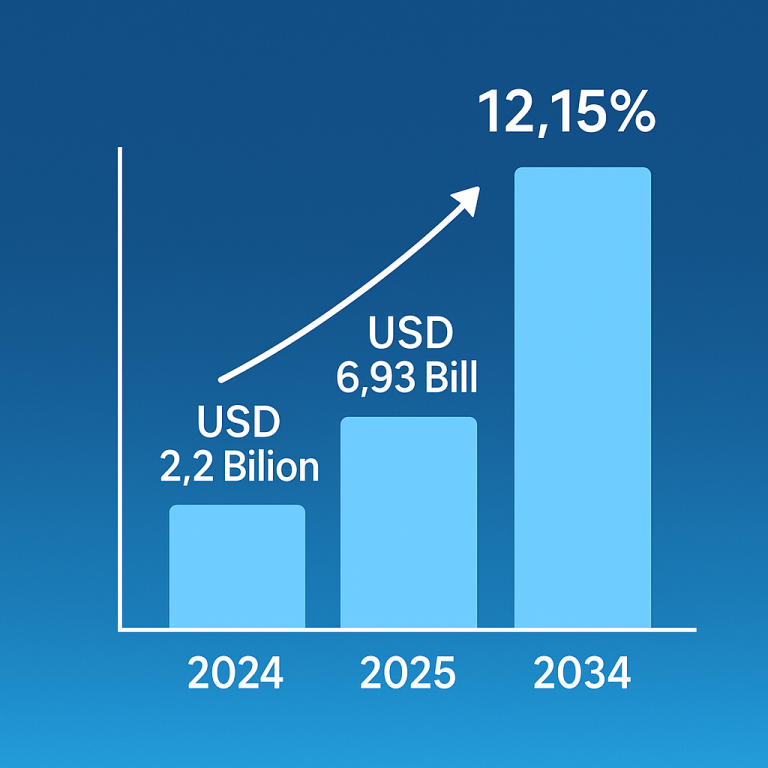
Disease-Modifying Therapy: A Beacon of Hope
Disease-modifying therapy emerges as a superhero in the HD treatment market. Unlike traditional symptom-focused treatments, it aims to alter the course of the disease itself. Imagine a therapy that not only alleviates symptoms but also slows or halts the progression of HD. This transformative potential sparks hope and increases demand for such therapies.
The pursuit of disease-modifying therapy not only improves patient outlook but also fuels enthusiasm among pharmaceutical companies, researchers, and healthcare providers. The focus shifts from symptom management to making a substantial impact on the lives of those dealing with HD. The demand for these game-changing therapies propels the market forward, fostering optimism for a future where HD can be effectively managed and, ideally, halted in its tracks.
Huntington’s disease (HD) poses a unique challenge as it is primarily inherited, transferring from parent to child through a gene mutation. The risk of developing Huntington’s disease is intricately tied to genetic factors, and understanding these dynamics is crucial.
For any specific customization click here: https://www.towardshealthcare.com/personalized-scope/5100
Inherited Risk: Familial Huntington’s Disease
When a parent has Huntington’s disease, the transmission to their child occurs through a specific mutation on chromosome 4. Each child of an affected parent faces a 50% chance of inheriting the chromosome carrying the HD mutation. If the child does not inherit this mutation, they are not at risk of developing HD and cannot pass it on to subsequent generations.
Sporadic Huntington’s Disease: Uncharted Genetic Waters
In instances where Huntington’s disease manifests without a family history, it is termed sporadic Huntington’s disease (HD). This form is caused by a mutation in the huntingtin gene, resulting in an abnormal repetition of DNA building blocks known as CAG repeats.
- CAG Repeats and Risk Levels:
- Individuals with fewer than 27 CAG repeats generally face minimal risk and are unlikely to develop HD.
- Those with repeats in the middle range (27 to 35) may not likely develop the disease themselves but could potentially pass it on to their offspring.
- Individuals with 36 or more CAG repeats are more predisposed to Huntington’s disease.
The 50% Rule: Genetic Odds for Offspring
Children of a parent with HD confront a 50% chance of inheriting the HD gene. If they do not inherit it, they will not develop HD and typically will not be carriers to pass it on to future generations. It’s essential to approach these genetic factors with compassion and a comprehensive understanding.
Huntington’s Disease: A Genetic Disorder with Multifaceted Effects
HD manifests as a genetic disorder impacting the brain, leading to movement problems, cognitive decline, and emotional challenges. While symptom management involves medications and support, the complexity of the condition persists.
Towards Innovative Solutions: Exploring Gene Therapies
In the pursuit of effective treatments, innovative solutions such as gene therapies are under exploration. However, fully controlling Huntington’s disease remains a formidable task, necessitating a dedicated focus on ongoing research and treatment advancements. As scientists delve deeper into the intricacies of the genetic landscape, hope emerges for enhanced interventions and improved quality of life for those affected by this complex disorder.
In 2022, the prevalence of Huntington’s disease (HD) worldwide underscores the urgent need for intensified research and advanced treatment options. According to the Journal of Neurology Research, an estimated 2.7 out of every 100,000 people globally are living with HD. However, regional variations exist, with higher prevalence observed in countries such as Canada, the United States, the United Kingdom, and Australia, compared to Asian nations like Japan, Korea, Taiwan, and Hong Kong. Notably, British Columbia, Canada, reports a prevalence of 13.7 per 100,000 individuals, slightly lower than the general Caucasian population.
Unraveling the Genetic Complexity of HD
HD is inherited through a mutated gene, where possessing one copy from either parent can lead to the disease. The expansion of a specific DNA sequence in the HD gene is the culprit. This gene instructs the production of huntingtin, a protein prevalent in the central nervous system. Though the exact function of huntingtin remains elusive, it is believed to play vital roles in various cellular processes.
Symptoms of HD typically manifest when the repeated DNA sequence reaches 36 or more, with full-blown symptoms often observed at 40 repeats or above. The number of repeats correlates with an earlier onset, faster progression, and increased severity of the disease.
Precision Challenge in Therapeutic Development
HD poses a unique challenge in therapeutic development due to its genetic nature. Crafting treatments that precisely target the mutated gene responsible for HD while sparing healthy genes presents a formidable puzzle. It’s akin to fixing a glitch in a massive computer program without unintended side effects, where each gene plays a vital role in a well-orchestrated symphony.
The precision required in therapeutic interventions resembles delicate molecular surgery. Any lack of precision could inadvertently affect healthy genes, complicating the landscape. This complexity in targeting the root cause hinders the development of effective therapies, impacting the market for HD treatments.
Read More Highlights: